Cahan Lab  Johns Hopkins University
Johns Hopkins University
Resources
Education and Presentations
Learn about mechanisms underpinning multipotency and self-renewal of stem cells. Emphasis on seminal studies and bleeding edge technologies, and the critical contributions of computational approaches to both. Homeworks involve analysis of real single cell omics data and final project requires development of novel analysis approaches in Python.
Spring 2025 Course website
Spring 2024 Course syllabus and companion website
Spring 2023 Syllabus on Canvas
Spring 2022 Course website
Spring 2021 Syllabus
Spring 2020 Syllabus
Spring 2019 Syllabus
 Academic Job Search Seminar
Academic Job Search Seminar
Part of a series of seminars to demystify the process of applying for academic positions. My presentations cover a lot of 'nuts and bolts' details. The first presentation has tips on the application process, the second covers the interview(s).
The Application (Apr 2016)
The Interview (Sept 2016)
Code, Data, Web Applications
Each box below points to resources associated with one of our papers. Some papers describe new computational methods, some papers describe omic data, and some papers describe both new computational methods and new omic data. Some boxes have links to web applications that we created to broaden accessibility to our data or methods. More recently, we have started to create repositories that contain the analyses steps required to reproduce the results in our papers.
Epigenetic memory of acinar-to-ductal metaplasia
DNA methylation memory of pancreatic acinar-ductal metaplasia transition state altering Kras-downstream PI3K and Rho GTPase signaling in the absence of Kras mutation
Paper: Lo et al, Genome Medicine 2025
Data: GSE249143
DNA methylation memory of pancreatic acinar-ductal metaplasia transition state altering Kras-downstream PI3K and Rho GTPase signaling in the absence of Kras mutation
Paper: Lo et al, Genome Medicine 2025
Data: GSE249143
OneSC is a computational platform that simulates synthetic cells across pseudotemporal trajectories using systems of stochastic differential equations governed by a gene regulatory network (GRN). After you give OneSC your scRNA-seq data of a temporal process, it will infer a GRN and then allow you explore how perturbations impact the trajectories and final state composition of the system. For example, you can use OneSC to perform in silico knock-down or over-expression experiments.
Paper: Peng D and Cahan P, Bioinformatics. 2024
Code: OneSC on GitHub
Documentation: OneSC ReadTheDocs
StemFinder
stemFinder predicts single cell differentiation time based on heterogeneity in cell cycle gene expression. Written in R.
Paper: Noller K and Cahan P, Brief Bioinfo. 2024
Code: stemFinder code on GitHub
Scripts to reproduce paper results: Scripts on GitHub
stemFinder predicts single cell differentiation time based on heterogeneity in cell cycle gene expression. Written in R.
Paper: Noller K and Cahan P, Brief Bioinfo. 2024
Code: stemFinder code on GitHub
Scripts to reproduce paper results: Scripts on GitHub
Direct comparison of mass cytometry and single-cell RNA sequencing of human peripheral blood mononuclear cells
Paper: Su et al, Scientific Data 2024
Data:
scRNA-seq of Drosophila embryos at stages 10-12 (20,585 cells) and stages 13-16 (42,727 cells). There are four samples in total.
Paper: Peng et al, Development 2023
Analysis: https://github.com/CahanLab/fruitfly_organogenesis
Data: scRNA-seq of Drosophila embryo, stages 10-12 and 13-16
PACNet quantifies transcriptional fidelity of engineered cells from bulk RNA-Seq data. It enables benchmarking new cell fate engineering protocols against current standard methods based.
Paper: Lo et al, Stem Cell Reports 2023
Code: PACNet repo
Web App: PACNet web app
Epoch infers gene regulatory networks (GRNs) that are dynamic, in that their topologies change over time, from scRNA-seq data. It is fast and performs well based on the Beeline GRN benchmarking platform.
Paper: Su, Spangler et al, Stem Cell Reports, 2022
Code: Epoch in R and Epoch in Python
Data: scRNA-seq of mouse embryoid body differentiation, day 0 to day 4
 PySCN
PySCN
PySCN enables comparions of embryo models such as embryoids, gastruloids, embryoid bodies, to in vivo embryos with scRNA-seq. PySCN includes curated reference data, and it allows the user to perform classification and enrichment analysis with curated, development-specific gene signatures.
Paper: Tan et al, bioRxiv 2021
Code: PySCN code and PySCN documentation
Analysis: https://github.com/CahanLab/vivo_vs_vitro
Web App: SingleCellNet Web App
CancerCellNet measures the transcriptional similarity of cancer models to 22 naturally occurring tumor types and 36 subtypes, in a platform and species agnostic manner.
Paper: Peng, et al, Genome Medicine, 2021
Code: https://github.com/CahanLab/cancerCellNet
scRNA-seq of 7,329 synovial joint progenitor cells (Gdf5-lineage) from the mouse hindlimb knee from E12.5 to E15.5. There is one sample per timepoint.
Paper: Bian et al, Development, 2020
Web app: Web interface to scRNA-seq data
Data: scRNA-seq of Gdf5-lineage cells
SingleCellNet is a computational tool that classifies scRNA-Seq data across platforms and across species. It transforms query and reference data with the top-scoring pair, and then uses a Random forest for classification.
Paper: Tan et al, Cell Systems, 2019
Code: SingleCellNet code in R
Documentation: https://cahanlab.github.io/singleCellNet/
 Embryoid body transcriptional states
Embryoid body transcriptional states
In this paper, we described the transcriptional states in mouse embryoid body differentiation at bulk and single cell levels. We generated bulk RNA-seq for day 0, 2, 4, and 6. And we generated scRNA-seq for days 4 and 6.
Paper: Spangler, et al, Stem Cell Research, 2018
Data: GEO accession for both bulk and scRNA-seq
A protocol for applying the CellNet method to bulk RNA-seq data. This protocol starts with raw sequencing reads so that they are processed in the same way as the training data.
Paper: Radley, et al, Nature Protocols, 2017
Code: https://github.com/CahanLab/CellNet
 Deconstructing transcriptional heterogeneity in pluripotent stem cells
Deconstructing transcriptional heterogeneity in pluripotent stem cells
We used single-cell molecular profiling of mouse pluripotent stem cells subjected to a range of perturbatio factors to infer mechanisms that contribute to pluripotency and self-renewal.
Paper: Kumar, Cahan, et al, Nature, 2014
Data: GEO accession of scRNA-seq and bulk ChIP-Seq.
 CellNet: Network Biology Applied to Stem Cell Engineering
CellNet: Network Biology Applied to Stem Cell Engineering
We designed a network biology computational method (CellNet) to assess the fidelity of cell fate engineering and to generate hypotheses for improving cell fate engineering protocols.
Paper: Cahan, Li, Morris, et al, Cell, 2014
Code: Original CellNet code
Documentation: https://pcahan1.github.io/cellnetr/
Dissecting Engineered Cell Types and Enhancing Cell Fate Conversion via CellNet
We used CellNet-predictions to improve B cell to macrophage direct conversion. CellNet also uncovered an unanticipated intestinal program in induced hepatocytes (iHeps), validated by long-term functional engraftment of mouse colon by iHeps.
Paper: Morris, Cahan, Li, et al, Cell, 2014
Data: GSE59037
 Transcriptional landscape of hematopoietic stem cell ontogeny
Transcriptional landscape of hematopoietic stem cell ontogeny
Transcriptional profiling of hematopoietic progenitors from the AGM, Placenta, Yolk Sac, fetal liver and bone marrow, as well as ESC-derived hematopoietic stem-like cells. Cells were sorted with surface markers that enrich functionally for hematopoietic repopulation.
Paper: McKinney-Freeman, Cahan, Li, et al, Cell Stem Cell, 2012
Data: GEO accession to microarray data
Web app: StemSite: web interface to expression data and transcriptional modules
 The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells.
The impact of copy number variation on local gene expression in mouse hematopoietic stem and progenitor cells.
We used high-density aCGH to measure DNA copy number variation in the genomes of 19 commonly used inbred mouse strains. Based on association between CNV occurrence and expression in cis, we estimate that up to 28% of strain-dependent expression variation is associated with copy number variation in hematopoietic stem and progenitor cells.
Paper: Cahan, et al, Nature Genetics, 2009
Data: aCGH DNA copy number data and microarray expression data
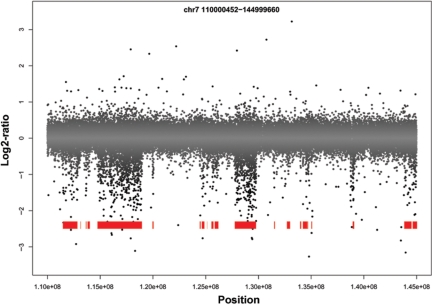 wuHMM
wuHMM
We invented wuHMM, an algorithm based on Hidden Markov Models for calling DNA copy number variants from array comparative genomic hybridization (aCGH) data. wuHMM takes advantage of SNP data to infer regions of high sequence divergence to reduce the false positives.
Paper: Cahan, et al, Nucleic Acids Research, 2008
Code: https://github.com/CahanLab/wuhmm
Data: GEO accession of the aCGH data used to test wuHMM